Im Jahr 2020 wurde erstmals über einen neuen, den autoinflammatorischen Syndromen (AI) zugeordneten Symptomkomplex berichtet, der als VEXAS-Syndrom bezeichnet wurde (Ref.1). AI Syndrome (AIS) sind Erkrankungen, die durch eine Überreaktion des angeborenen (Eng. innate) Immunsystems ohne Infektionen oder Bildung von Autoantikörpern bedingt sind. Man unterscheidet AIS mit genetischer Basis von solchen ohne bekannte genetische Grundlage. Die ersteren beruhen auf Mutationen, die zu einer Überreaktion von Inflammasomen führen (= zytosolische Multiproteinkomplexe des angeborenen Immunsystems, die zur Aktivierung von Entzündungsreaktionen führen). Dazu zählen z.B. Mutationen, die für das familiäre Mediterrane Fieber (FMF) oder die Gruppe der Cryopyrin-assoziierten, periodischen Syndrome (CAPS) verantwortlich sind. Zu den AIS gehörten auch jene, die durch Adjuvantien von Impfstoffen oder durch Fremdkörperimplantate (z.B. Silikon-Brustimplantate) hervorgerufen werden. Diese Varianten von AIS werden als ASIA (Autoimmune Inflammatory Symdrome induced by Adjuvants) bezeichnet.
Das VEXAS-Syndrom ist ein sehr seltenes, angeborenes, aber erst im Erwachsenenalter manifestiertes Syndrom (Ref. 2), das nur das männliche Geschlecht betrifft. Es zählt zu den genetisch determinierten AIS.
Eine Mutation von UBA1 in den Keimzellen ist bereits von der Geburt letal. VEXAS ist jedoch durch somatische Mutationen an der Position 41/Methionin des UBA1 Gens bedingt, das für das E1-Enzym codiert. Dieses Enzym ist für die Aktivierung des Prozesses der Ubiquitierung verantwortlich. Ubiquitin ist ein kleines Protein, das in allen enkaryoten Zellen vorkommt (also überall-ubiquitär-vorhanden ist) und eine zentrale Rolle beim Abbau von zellulären Proteinen spielt. Mit Ubiquitin verbunde Proteine (=Ubiquitierung) werden in den Proteaosomen degradiert. UBA1 ist am X-Chromosom lokalisiert, und die Mutationen manifestieren sich ausschliesslich in Zellen der myeloischen Reihe. Stammzellen anderer hämatopoietischer Linien bleiben intakt, weil die betroffenen Zellen (z.B. T-Zellen) im Knochenmark durch einen noch nicht geklärten Prozess eliminiert (=negativ selektioniert) werden. Der durch UBA1 Mutationen bedingte, gestörte Proteinabbau in den betroffenen Zellen resultiert in der Entwicklung verschiedener, sich überlappender Entzündugnsprozesse. Ursache ist die Expression einer neuen, katalytisch funktionsgestörten Isoform des E1-Enzyms in den Zellen.
Klinik
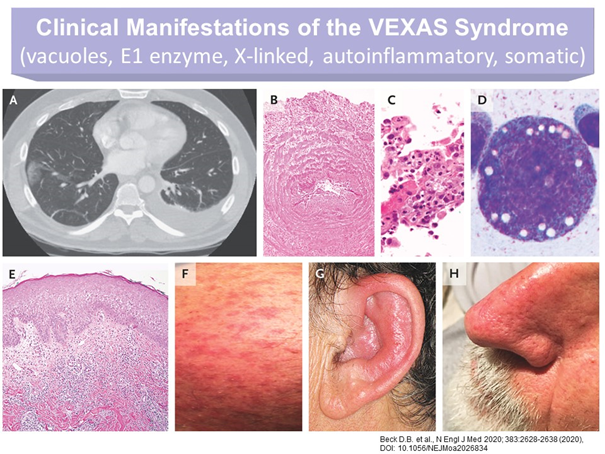
Die klinischen, therapeutisch nicht signifikant beeinflussbaren Symptome des VEXAS-Syndroms sind sehr heterogen. Sie treten, wie erwähnt, erst im späteren Erwachsenenalter auf und umfassen (Abb.1):
- Rekurrierendes Fieber
- Knochenmarksdysplasie
- Zytopenie mit charakteristischen Vakuolen in myeloischen und erythropoietischen Vorläuferzellen
- Neutrophile Entzündungen in Haut und Lunge
- Polychondritis
- Vasculitiden
Bei den 25 Patienten in der Erstbeschreibung (Ref.1) und bei späteren Fallberichten (z.B. Refs.3, 4) zeigte sich eine Kombination mehrerer Entzündungssyndrome, wie relapsierende Polychrondritis (v.a. Ohr und Nase), Sweet-Syndrom (= akute,febrile, neutrophile Dermatose), Polyarthritis nodosa, Riesenzellarteriitis, hämatologische Erkrankungen (myelodysplastisches Syndrom, makrozytäre Anämie, multiples Myelom) und Thromboembolismen.
Für eine genaue Abschätzung von Inzidenz und Prävalenz ist der Zeitraum seit der Erstbeschreibung noch zu kurz.
Immunologische Charakteristika
- Aktivierung mehrerer proinflammatorischer Signaltransduktionswege in myeloischen Zellen des angeborenen Immunsystems, manifestiert durch erhöhte Ausschüttung proinflammatorischer Zytokine (TNFα, IL-6, IL-8, IFNγ)
- Gestörte Ubiquitierung in myeloischen Zellen
- Vermehrte Neutrophilenaktivierung, manifestiert durch verstärkte Netzbildung (=NETosis)
- Keine Autoantikörper
Diagnostik
- Erhöhte Blutsenkung
- Erhöhtes CRP, Fibrinogen und D-Dimer
- Bisher publizierte unspezifische Parameter, die für eine Aktivierung des angeborenen Immunsystems sprechen (proinflammatorische Zytokine s.o.)
Genetik
Abb. 1:
Referenzen:
Ref. 1:
Beck, D. B., et al., (2020). Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease. New England Journal of Medicine, 383(27), 2628–2638. https://doi.org/10.1056/nejmoa2026834
Ref. 2:
Cantarini, L., Lucherini, O. M., & Rigante, D. (2013). Caution Should be Used in the Recognition of Adult-Onset Autoinflammatory Disorders: Facts or Fiction? Frontiers in Immunology, 4. https://doi.org/10.3389/fimmu.2013.00096
Ref. 3:
Lötscher F, et al., (2022) Case Report: Genetic Double Strike: VEXAS and TET2-Positive Myelodysplastic Syndrome in a Patient With Long-Standing Refractory Autoinflammatory Disease, Front. Immunol. 12:800149, doi: 10.3389/fimmu.2021.800149
Ref. 4:
Grayson P.C., et al. (2022), Case 2-2022: A 70-year-old man with a recurrent left pleural effusion, N Engl J Med 2022; 386:274-283, DOI: 10.1056/NEJMcpc2115847